gRNA Design
SPEED
Fast and easy gRNA design using ATUM’s design tool that minimizes off-target effects using ATUM’s scoring algorithms. ATUM will clone your gRNAs into the CRISPR construct of your choice and send you ready-to-transfect plasmid.
SPECIFICITY
Easy design of two tandem gRNAs for NickaseNinja vectors to enhance specificity.
PRECISION
The design tool enables <br>suppression of off-target effects.
gRNA Design
gRNA Design Overview
Design gRNA(s) to efficiently engineer your target and minimize off-target effects by using ATUM’s Scoring Algorithm.
The ATUM gRNA Design Tool enables:
- Design gRNAs for wild-type or Nickase Cas9 vectors
- Easy design of 2 tandem gRNAs for NickaseNinja vectors
- Fast design against gene name, locus or specific target sequence
- Suppression of off-target effects by avoiding sequences that match elsewhere in the genome
- Direct ordering of your selected gRNAs in Electra CRISPR vectors, Transfection Ready
Enter your target gene name or your target sequence, and the design tool will identify all Cas9 target sites within the input sequence. The results contain a rank ordered list of target sites based on predicted specificity. The algorithm used in the program is based on the occurrence of the 12 base pair seed sequence preceding the NGG and NAG protospacer-adjacent motif (PAM).

ATUM CRISPR gRNA Design Tool Screenshot. The design tool generates gRNAs in rank order coded by specificity allows you to visualize the position for your gRNA relative to the splice variants and any overlapping genes. It is routine in published literature to test more than one gRNA to maximize likelihood of success.
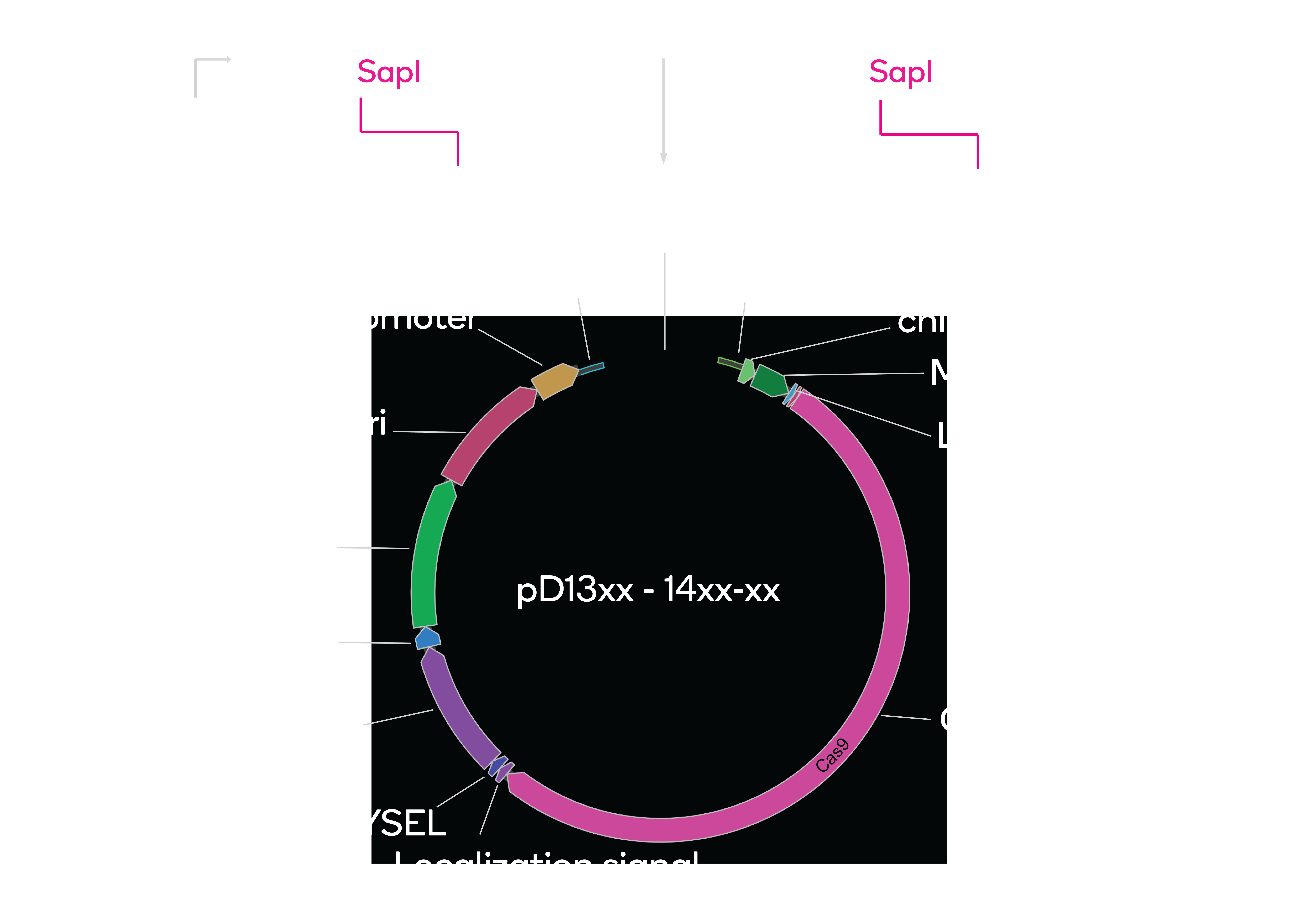
PCR your gRNA
To PCR your gRNA, we recommend you add the following ends to your primers, as these contain the Electra sites to clone directly into pDAUGHTER-CRISPR vectors.
Forward primer:
5′-TACACGTACTTAGTCGCTGAAGCTCTTCTCCG….(gRNA)….-3′
Reverse primer:
5′-AGGTACGAACTCGATTGACGGCTCTTCTAAC….(gRNA Reverse Complement)….-3′
You can either anneal your primers directly or design primers with 15-20 bp overlaps with the recommended ends and “anneal and extend”. We recommend 10 cycles of PCR. Use 1µl of PCR reaction in the Electra reaction shown below.
* Your gRNA must not contain any SapI recognition sites, since the Electra cloning process utilizes the type IIS enzyme SapI.
Protocol

*Electra Cloning Kit reagents (Catalog #EKT-03)
- Combine components as listed in table above in a single 1.5ml tube. Note Daughter Vectors come pre-linearized from ATUM.
- Incubate 5 – 20 minutes at room temperature.
- Transform 2 µl of each reaction into competent cells.
- Plate onto LB plates with selection antibiotic.
- Incubate overnight at 37°C. Pick transformants.
What can I do with CRISPR/Cas9 technology?
Use CRISPR Cas9 to knockdown gene expression, via indel mutations, and to knock-in genes at precise locations in genomes of your choice.
Can I use CRISPR/Cas9 technology in my host?
We currently have developed products for mammalian and yeast systems. We have ongoing studies to develop CRISPR in E.coli and additional host systems, many through collaborations. We would love to work with you to develop CRISPR/Cas9 to work in your system. Please contact us at communications@atum.bio.
Can I get a custom CRISPR vector?
Yes, you can access our expertise to create a customized CRISPR vector, ideal for your specific host. Learn more about Custom CRISPR Vectors.
What is NickaseNinja?
NickaseNinja is an all in one vector that expresses the nickase mutant of Cas9 along with 2 gRNAs that are necessary for the protein to function.
How do I order NickaseNinja?
The NickaseNinja is currently only offered with our cloning services. We can clone your custom designed gRNAs into the vector for you.
How does the ATUM gRNA Design Tool work?
Enter your gene or sequence of interest and the design tool will identify all Cas9 target sites within the input sequence. The results contain a rank ordered list of target sites based on predicted specificity. The algorithm used in the program is based on the occurrence of the 12 base pair seed sequence preceding the NGG and NAG protospacer-adjacent motif (PAM).
What is Indel Frequency?
Indel frequency is a measure of the insertion and/or deletion rate of the target gene sequence. Indels are small insertions or deletions at the target site, presumably due to cutting by cas9/gRNA and then non-homologous end joining (NHEJ) in the host cell. NHEJ introduces small mutations (insertions or deletions, indels) that then escape cutting by Cas9/gRNA. After exposure to cas9/gRNA, we PCR out the region of interest for 12-24 individual colonies and sequence through the cut site. We then calculate the ratio of mutation or insertion/deletion at the cut site (indels) to unmodified wildtype sequence (wildtype) to get indel frequence = indels/wildtype*100 (as a percentage).
What types of Indels are typically observed?
The most common is a 1bp deletion. We’ve seen 1-10bp deletions as well as small insertions. In the literature we see reports of between 1-10bp insertions and deletions.
What is the maximum insertion size of new DNA sequences?
Depends on the host. The maximum size of insertion in a particular host probably doesn’t change (it’s whatever is described in the literature for that host). CRISPR simply makes it more efficient, so you can do this without selection markers. You may be able to increase the maximum insert size slightly since the process is now more efficient, but you’ll have to experimentally validate this in your host.
How do you select for positive cells?
CRISPR is so efficient that you don’t need selection. We usually pick 12-24 clones to propagate and screen by sequencing. Usually at least 1 in 10 will contain the appropriate frameshift mutation that results in a gene knockout. You can use antibiotic markers if you like, but CRISPR is so efficient that marker-less genome editing is common and usually the desired approach with CRISPR.